Tag: Lab
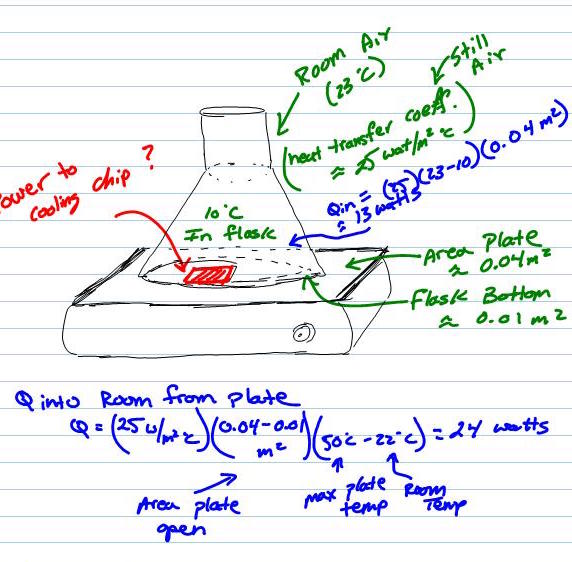
Cooling A Flask?
So, you have a flask (maybe it contains some chemical) and you want to cool the flask. The obvious answer is to place it in a refrigerator. However, you want to keep it on a magnetic stir plate, which every well stocked kitchen has on hand. You do have a few options, the first is…
Read More
Publication – Lowering the cost of cellulase
There is a new publication that was just accepted for print regarding the production of cellulase. This article is entitled “Nutrient control for stationary phase cellulase production in Trichoderma reesei Rut C-30″ and will appear in the journal Enzyme and Microbial Technology. Cellulase is normally produced by a fungus, Trichoderma ressei. Most of the time the cells continually grow…
Read More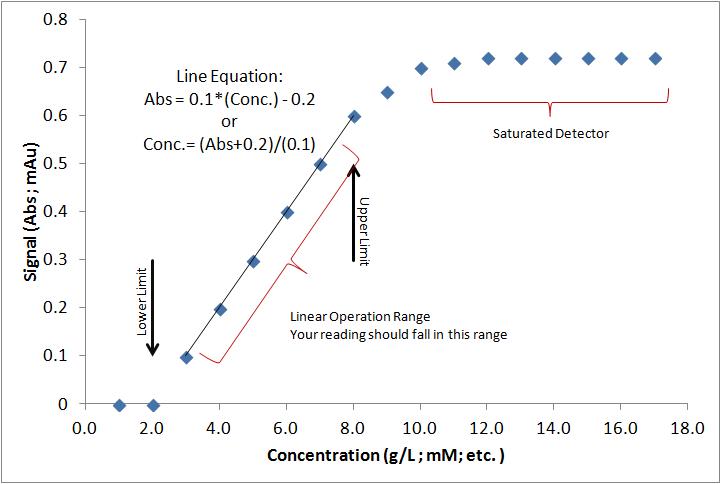
Calibration Data
A question that I seem to receive frequently: “Can you help me with the calibration?” So, you have a sample. And, there is some chemical in there (like glucose) and you want to know how much glucose is in the sample… maybe you have some idea, it should be less than something, right? Were do…
Read More